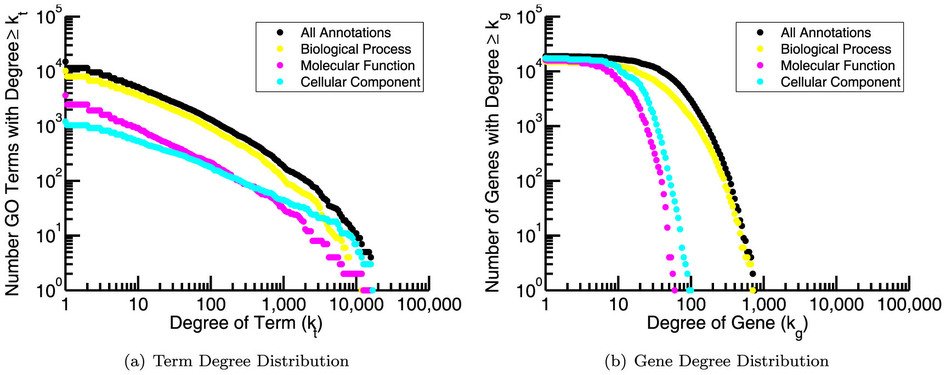
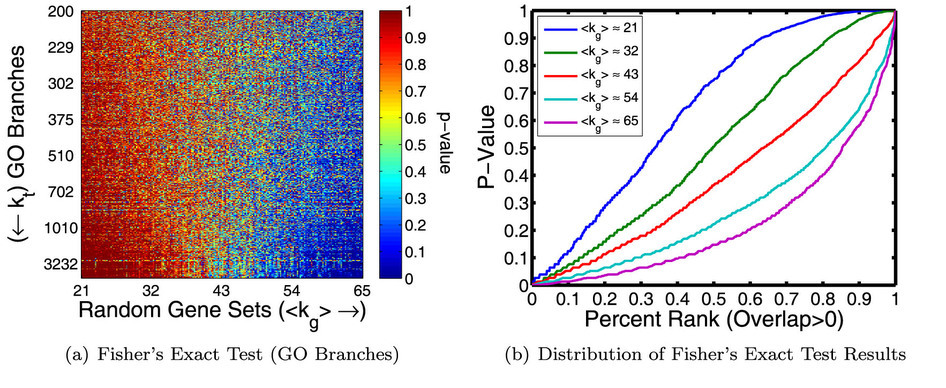
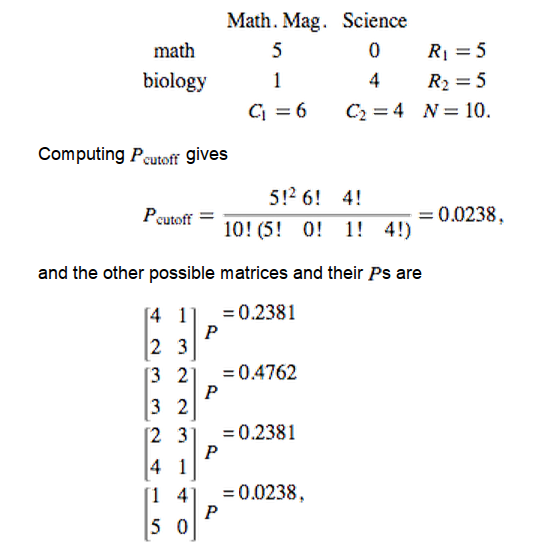Overview
- Recent high-throughput methods (microarray, RNA-Seq, etc) made it easy to produce large datasets comparing samples in different conditions.
- The end result of many of these analyses, however, is often a large list of genes that are associated with one condition or the other.
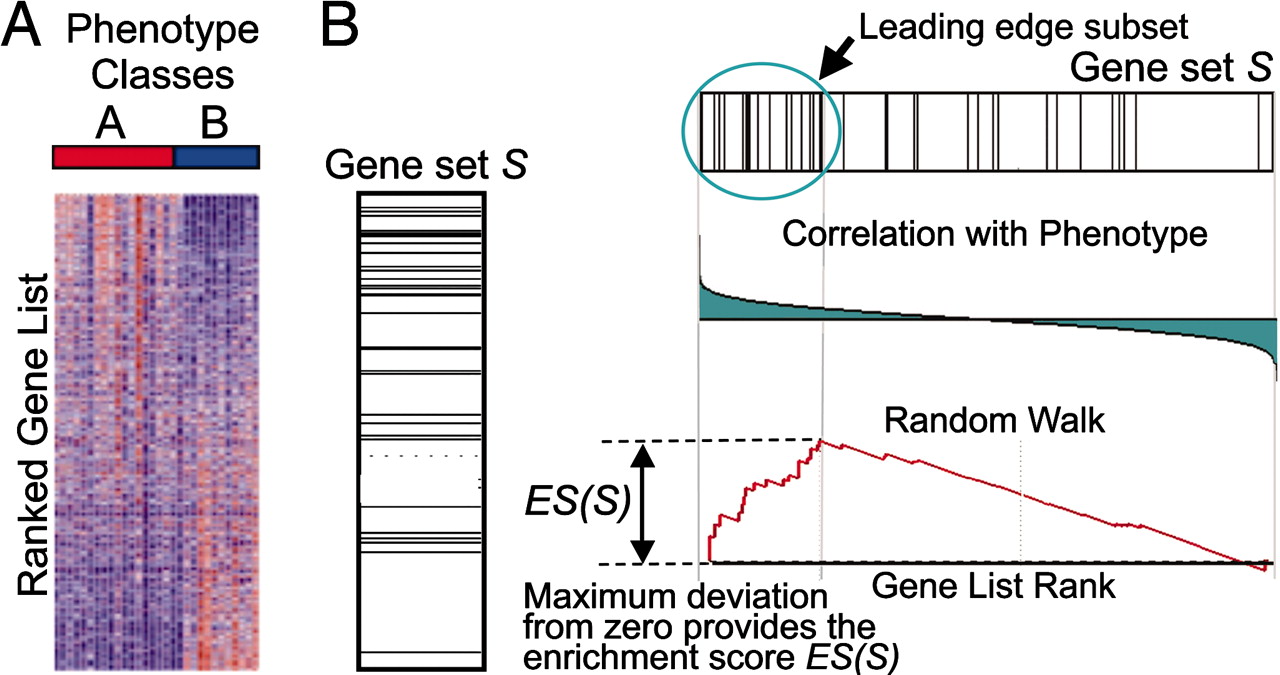
- Numerous tools have been developed to look for "enrichment" in these resulting gene sets for genes associated with a particular known pathway or functional annotation.
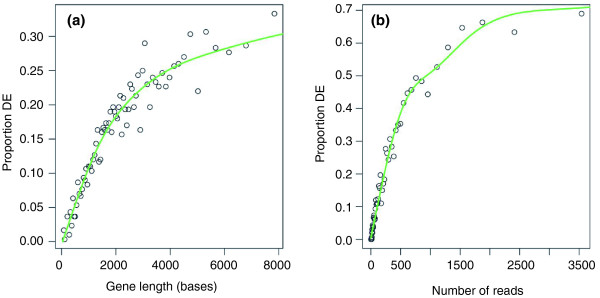
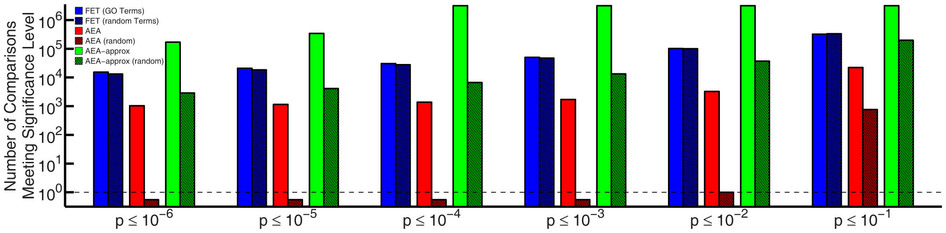
- These methods (GSEA, etc) often use statistics which make some assumptions about the distribution of annotations which may not be valid.
- What are the effects of these assumptions the resulting interpretation?
- Can we do better?